

Abstract
Non-stoichiometry and bulk cation transport have been identified as key factors in the release and uptake of hydrogen in the Li–N–H system. Amide halide phases have been synthesized that have ionic conductivities several orders of magnitude greater than lithium amide, a faster rate of hydrogen release and elimination of the by-product, ammonia. Here we report the effect of both anion- and cation-doping on the hydrogen desorption properties of lithium amide, focusing in particular on how the presence of chloride anions and magnesium cations affects and controls the structure of the amide and imide compounds at the sub-nanometre level. Reducing the chloride content resulted in new low-chloride rhombohedral phases that contain around half of the chloride present in earlier amide chlorides, but maintained the enhancements seen in hydrogen desorption properties when compared to the halide-free system. These materials may also have potential in a range of other energy applications such as all solid state lithium ion batteries, supercapacitors, and CO2 capture and storage membranes.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
The transition to a sustainable, environmentally benign, carbon-free energy system that can meet our ever-increasing demands is arguably the defining challenge of our age [1]. A key deficiency is the continuing lack of a convenient, efficient and safe means of storing renewable energy, in particular for mobile applications, with electrochemical—in the form of lithium ion batteries—and chemical storage—in the form of hydrogen gas—considered the most promising solutions.
In automotive transport the move away from fossil fuels towards electric vehicles has already begun [2], but energy density and cost issues severely limit the range of battery-only cars, a problem exacerbated by long recharge times [3]. Fuel cells powered by hydrogen offer much higher energy density and shorter refuel times, and may either act as a range-extender alongside [2]—or replace [3]—batteries in electric vehicles. Hydrogen is an ideal fuel as it has a far higher specific calorific value than any other (2.6 times that of petrol), and is virtually emission/pollution free at the point of use. In addition, the ease of interconversion of electricity and hydrogen as energy carriers potentially provides a solution to the issue of intermittency, in principle rendering the gas a key component of a renewable-energy system [3]. Yet proponents and opponents alike, of the use of hydrogen as a green energy carrier, are agreed that the absence of a low-cost, safe and convenient means of storage represents a serious impediment to its wider use.
Although many complex hydrides can reversibly store large amounts of hydrogen [4], even in those cases where the thermodynamic properties are deemed favourable for reversible hydrogen release under moderate conditions, the kinetics of desorption and rehydrogenation of light metal complex hydrides remain too slow for them to be used as practical hydrogen storage materials. Nanostructuring, in particular through the addition of transition metal catalysts, mechanically milled into the hydride to achieve dispersion and hence accompanied by the benefits of refinement of particle and grain size, is known to improve the hydrogen de/reabsorption kinetics of many hydrides by facilitating splitting of the hydrogen molecule and helping hydrogen to diffuse through passivated surface layers or along grain boundaries in certain cases. Much of the current interest in lightweight complex hydrides was sparked by the discovery of Bogdanović and Schwickardi [5] that the addition of TiCl3 significantly improved the reversibility of NaAlH4. This classic example remains a benchmark for complex hydrides, but although the mechanisms of the desorption/hydrogenation processes in this system, and of how the catalysts improve reaction rates, have attracted more scrutiny than most, these studies have struggled to improve on rehydrogenation over many hours, under 160 bar at 200 °C.
One fundamental area that has received comparatively little attention to date is rates of diffusion within the bulk materials themselves. As recent work [6, 7] is now beginning to make clear, this represents a significant oversight. The reaction of lithium amide with lithium hydride (LiH) is known [8, 9] to release 10.4 wt% of hydrogen in two steps:
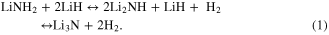
Step 1 has been shown to proceed via the release of ammonia (NH3), which quickly reacts with lithium hydride to produce hydrogen [10]. A reduction in temperature of H2 release may be achieved by replacing the lithium amide by its magnesium counterpart [11], which releases ammonia at a lower temperature. Similar lithium magnesium imide products are formed through the reaction of lithium amide and magnesium hydride, but these are found to rehydrogenate preferentially back to magnesium amide and lithium hydride. Step 2, forming lithium nitride, is often discounted as a hydrogen storage reaction, as complete desorption in dynamic vacuum requires temperatures in excess of 320 °C.
Experiment [12] and theory [13] have highlighted lithium ion mobility as a pivotal factor in both reactions. The close structural relationship between LiNH2 and Li2NH may allow for a topotactic de/rehydrogenation via a solid solution of the form Li1+nNH2−n. The bulk structural transformation occurring in these reactions equates to the diffusion of Li+ into and H+ out of (or vice versa) the face centred cubic anion lattice. Li2NH has an isotropic room temperature ionic conductivity of 3 × 10−4 S cm−1, increasing to 2 × 10−2 S cm−1 at 100 °C, with an activation energy of 56 kJ mol−1 [14]. Li3N has a very high anisotropic room temperature conductivity of ~10−3 S cm−1, but its use in battery applications is limited owing to a low theoretical decomposition potential of 0.445 V [15]. In contrast, LiNH2 displays extremely low room temperature conductivity (< 10−8 S cm−1), increasing to 5 × 10−6 S cm−1 at 127 °C [16], suggesting that reduced ionic mobility in hydrogen-rich compositions could both slow hydrogen release from LiNH2 and impede full bulk rehydrogenation.
Materials chemists have long used cation and/or anion doping to improve properties in a wide range of different materials. Both were exploited in our preliminary study [6], which found a large range of new amide halide [Li1+m(NH2)Xm and LiMgn(NH2)X2n, where X = Cl, Br, I] and imide halide [Li2+m(NH)Xm and Li2Mgn(NH)X2n] phases that released hydrogen reversibly via reactions (2) and (3):
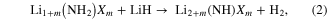
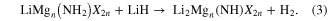
The most conducting of these, with room temperature ionic conductivities approaching 10−3 S cm−1, were also quickest to release hydrogen on heating and to hydrogenate, and unwanted ammonia release was suppressed. Reductions in the decomposition temperature of lithium amide caused by chlorides have subsequently also been reported by other workers [17, 18]. This marks ionic conductivity as potentially a crucial parameter in optimizing the properties of hydrogen storage materials, whose influence is unlikely to be restricted to nitrogen-containing systems [6, 7].
The key to these improvements lies in the ability of halide anions to affect and control the structure of the amide and imide compounds at the sub-nanometre level. The different phases of the amide chloride have different lithium ion site occupancies and so investigations into their structure may yield important insights into their conductivity and hydrogen storage properties. However, the addition of the relatively heavy halide anions reduces the gravimetric hydrogen capacity of the materials. This study investigates the phase space of lithium amide chlorides with the aim of reducing this gravimetric penalty whilst maintaining the improvements in hydrogen storage properties.
2. Experimental and procedure
Lithium amide (Sigma–Aldrich, 95%) and lithium hydride (Sigma–Aldrich, 95%) were used without further purification. Anhydrous lithium chloride and magnesium chloride (Sigma–Aldrich, ≥98%) were dried at 300 °C under high vacuum (1 × 10−6 mbar) for 24 h prior to use. All manipulations were performed in an argon-atmosphere glove box. Amide halides were synthesized by grinding the appropriate halide with LiNH2 in the desired molar ratio, placing the reaction mixture into a quartz tube, and heating under an argon flow (1 bar) at the required temperature for the specified reaction time.
Powder x-ray diffraction (XRD) data were collected on a Bruker D8 advance diffractometer operating in transmission geometry with a Cu-Kα1 x-ray source. Samples were sealed from the atmosphere between two pieces of amorphous tape. Powder XRD data for structural investigations were collected on a Siemens D5000 diffractometer in capillary mode with a Cu-Kα1 x-ray source. Samples were sealed into polyimide capillaries, allowing for long data collection times without degradation. Powder synchrotron XRD data were collected on beamline I11 at the diamond light source, Oxfordshire [19]. Samples were sealed in borosilicate glass capillaries to prevent degradation during transportation and measurement. Powder XRD data were analysed using the computer programme Topas [20] and structure solution was performed using Rietveld refinement of candidate models.
Temperature-programmed desorption with mass spectrometry (TPD–MS) data were collected using a home-built TPD apparatus described previously [21], coupled to a quadrupole mass spectrometer (HPR-20, Hiden Analytical). Samples were heated at 2 °C min−1 to 400 °C and held at temperature for 1 h before cooling.
3. Results
3.1. Effect of reaction time on structure
The reaction between lithium amide and lithium chloride was carried out at 400 °C at the molar ratio 3NH2−:1Cl−. As observed previously [6], Li4(NH2)3Cl is the product of this reaction and, at this molar ratio, two phases were observed under different conditions: a rhombohedral phase with a hexagonal unit cell was formed after 1 h and a body centred cubic phase was formed after a 12 h reaction time. In the cubic phase, the lithium sites are fully occupied. In the rhombohedral phase, the lithium sites are only partly occupied, potentially providing the opportunity for higher lithium ion mobility. Figure 1 shows the powder XRD patterns of the two phases.

Figure 1. Powder x-ray diffraction patterns of the two phases of Li4(NH2)3Cl.
Download figure:
Standard image High-resolution imageA detailed time dependence study was carried out of reaction times between 1 and 12 h. For reaction times of six or more hours, the cubic I213 phase of Li4(NH2)3Cl was formed, with reaction times shorter than this favouring the formation of the rhombohedral  phase. Figure 2 plots the unit cell volumes observed for these phases against reaction time. For a given reaction time, there was considerable variation in observed volume between different experiments, but the overall trend was that increased reaction time resulted in a denser rhombohedral phase and less dense cubic phase.
phase. Figure 2 plots the unit cell volumes observed for these phases against reaction time. For a given reaction time, there was considerable variation in observed volume between different experiments, but the overall trend was that increased reaction time resulted in a denser rhombohedral phase and less dense cubic phase.
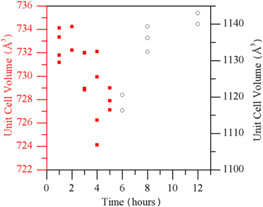
Figure 2. Unit cell volume against reaction time for the rhombohedral (red) and cubic (black) phases of Li4(NH2)3Cl; error bars for each point were much less than the variation between repeat experiments.
Download figure:
Standard image High-resolution imageThe transformation from the rhombohedral to the cubic phase was observed in real time using variable temperature powder synchrotron XRD. The rhombohedral phase was heated to 200 °C and then held at this temperature, with diffraction patterns recorded at regular time intervals (figure 3). The patterns show the gradual growth of peaks corresponding to the cubic phase and the corresponding reduction in intensity of those of the rhombohedral phase. The results of these experiments suggest that the cubic phase can be considered to be the thermodynamic product at this reactant ratio, with the rhombohedral phase favoured kinetically.

Figure 3. Powder synchrotron XRD patterns of Li4(NH2)3Cl after heating under different time and temperature conditions, showing the reduction in height of the peaks corresponding to the rhombohedral  phase (red, top right) and the appearance and growth of peaks representing the cubic I213 phase (black, bottom right).
phase (red, top right) and the appearance and growth of peaks representing the cubic I213 phase (black, bottom right).
Download figure:
Standard image High-resolution image3.2. Effect of chloride content on structure
The reaction shown in (4) was carried out over a range of Cl− compositions between x = 0.5 and 1.75 at 400 °C for reaction times of 1 and 12 h to investigate the effect of composition on the amide chloride under both kinetic and thermodynamic conditions
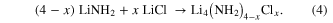
The phases observed across the range of x values are shown in figure 4. For x = 1, reaction for 1 h formed a rhombohedral  phase. Increasing the chloride content formed a cubic I213 phase, suggesting that the rhombohedral structure is unable to accommodate any additional Cl– ions into the structure beyond the NH2−:Cl− ratio of 3:1. This is likely to be because the structure of the rhombohedral phase requires Cl− ions to be located on the NH2− sites, which becomes disfavoured as the chloride content increases. When x was made more positive than 1.14, excess LiCl was observed in the XRD pattern in addition to the cubic phase; therefore additional Cl– ions cannot be accommodated into the cubic structure beyond the NH2−:Cl− ratio of 2.5:1.
phase. Increasing the chloride content formed a cubic I213 phase, suggesting that the rhombohedral structure is unable to accommodate any additional Cl– ions into the structure beyond the NH2−:Cl− ratio of 3:1. This is likely to be because the structure of the rhombohedral phase requires Cl− ions to be located on the NH2− sites, which becomes disfavoured as the chloride content increases. When x was made more positive than 1.14, excess LiCl was observed in the XRD pattern in addition to the cubic phase; therefore additional Cl– ions cannot be accommodated into the cubic structure beyond the NH2−:Cl− ratio of 2.5:1.
Figure 4. Phases present after a 1 h (top) and 12 h (bottom) reaction time plotted against x, where x is defined in equation (4).
Download figure:
Standard image High-resolution imageDecreasing the chloride content below x = 1 initially caused peak splitting in the XRD pattern, which can be refined as a mixture of two rhombohedral phases. Due to the presence of two phases, it is not possible to calculate the stoichiometries directly from the reactant ratios. Decreasing x below 0.7 caused excess LiNH2 also to be observed alongside the rhombohedral phases in the diffraction pattern of the reaction products.
The investigation into the 12 h reactions found that, as described previously [6], a cubic I213 phase is observed at x = 1. Increasing the Cl– content caused excess LiCl to be observed in addition to the cubic phase, suggesting no additional Cl– ions can be accommodated into the structure. As x was decreased below 1, a mixture of the cubic phase and an additional phase was observed until the new phase was observed alone at x = 0.57. This new phase can be refined to a hexagonal unit cell with rhombohedral symmetry, similar to the rhombohedral phases of both Li4(NH2)3Cl and Li7(NH2)6Br [6]. This x value corresponds to an NH2−:Cl− ratio of 6:1, which is in agreement with the structural refinement, implying a formula of Li7(NH2)6Cl for the new compound. At chloride contents lower than x = 0.57, excess lithium amide was observed, indicating that the lowest level of chloride that can be accommodated into the structure is a NH2−:Cl− ratio of 6:1.
3.3. Effect of chloride content on hydrogen desorption properties
As discussed previously, the amide chloride Li4(NH2)3Cl can accommodate a range of stoichiometry. However, this is not useful unless the improved hydrogen release properties are maintained, relative to LiNH2 + LiH. Reaction (5) was carried out for x values from 0.6 to 1 on samples formed after a 1 h reaction time
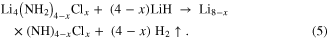
Temperature-programmed desorption measurements (figure 5) show that the onset and peak of hydrogen release remained constant for the x values where 0.6 ≤ x ≤ 1.
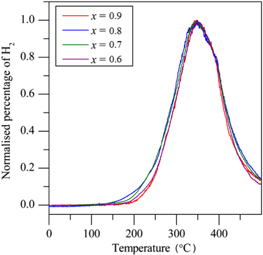
Figure 5. TPD–MS hydrogen desorption traces for  Clx + (4 − x) LiH; the traces have been normalized so that they may be more easily compared.
Clx + (4 − x) LiH; the traces have been normalized so that they may be more easily compared.
Download figure:
Standard image High-resolution imageThe structures of the imide chlorides formed as a product of reaction (5) were also investigated, to determine whether the non-stoichiometry was maintained in the dehydrogenated product. There was a decrease in the lattice parameter in the cubic cell of Li8−x(NH)4−xClx as x was decreased in the range 1 ≥ x ≥ 0.7, before it increased again between 0.7 ≥ x ≥ 0.6. This contraction of the unit cell, consistent with lower content of the larger chloride anion, can also be seen clearly on examination of the position of the (111) peak in the XRD patterns of these products. Figure 6 shows the shift of the (111) peak to a higher position as the value of x decreased, consistent with the decrease in unit cell size.
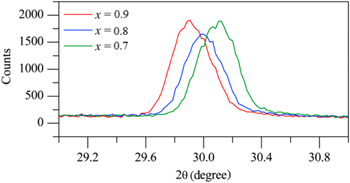
Figure 6. Powder x-ray diffraction patterns for the imide chlorides Li8−x(NH)4−xClx showing the (111) peak.
Download figure:
Standard image High-resolution image3.4. Lithium magnesium amide chloride
An equivalent phase space investigation was carried out on the mixed lithium magnesium system to see what effect reducing the chloride level had on the structure of the product of reaction (6) carried out at 400 °C for 9 h

As with the lithium only system, reducing the chloride level in the system caused a rhombohedral phase to be formed in preference to a cubic phase (figure 7). On reducing the chloride level to the NH2− to Cl− ratio of 6:1, a new phase was observed, with a structure the similar to that of the lithium-only rhombohedral phase observed at the same ratio but with a disordered replacement of one seventh of the Li+ ions with half as many Mg2+ (reaction (7))
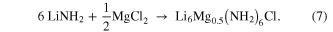
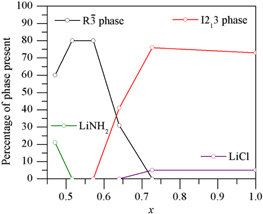
Figure 7. Phases present in the lithium magnesium system plotted against x, where x is defined in equation (6).
Download figure:
Standard image High-resolution imageReducing the chloride level below this ratio caused excess LiNH2 to be observed in the XRD patterns of the products, indicating that this is the lowest level of chloride that the structure can accommodate.
Temperature-programmed desorption measurements show that the onset and peak of hydrogen desorption were increased to a higher temperature as x was reduced from 1 to 0.57 for the lithium magnesium system (figure 8). Addition of magnesium reduced the temperature of desorption for the 3:1 NH2−:Cl− system, but had no effect on the 6:1 system. This difference may be understood as the lithium sites are fully occupied in the 3:1 structure, with the addition of magnesium ions causing vacancies to be created, compared to the 6:1 structure where vacancies are already present as the lithium ion sites are not fully occupied.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 8. TPD–MS hydrogen desorption traces for Li3Mg0.5(NH2)3Cl (black) and Li6Mg0.5(NH2)6Cl (red).
Download figure:
Standard image High-resolution image{kind=link}
The product formed from the reaction of Li3Mg0.5(NH2)3Cl with LiH was indexed to a cubic unit cell with a lattice parameter of a = 5.169(3) Å, although the peaks were relatively broad. This material was tentatively designated Li6Mg0.5(NH)3Cl. The product formed from the reaction of Li6Mg0.5(NH2)6Cl with LiH was a mixture of two cubic phases with a = 5.1233(6) Å and 5.1782(4) Å.
4. Conclusion
Non-stoichiometry and bulk cation transport have been identified as key factors in the release and uptake of hydrogen in the Li–N–H system [12]. Previously, we have reported that the addition of halides to lithium amide results in the formation of amide halide phases that have ionic conductivities several orders of magnitude greater than lithium amide, a faster rate of hydrogen release and elimination of the by-product, ammonia [6]. Here we have investigated further the effect of both anion- and cation-incorporation on the structural and hydrogen desorption properties of lithium amide, focusing in particular on how the presence of chloride anions and magnesium cations affects and controls the structure of the amide and imide compounds at the sub-nanometre level.
The reaction between lithium amide and lithium chloride at 400 °C in the molar ratio 3NH2−:1Cl− resulted in the formation of either a rhombohedral or cubic phase depending on the reaction time. The transformation from rhombohedral to cubic Li4(NH2)3Cl at 200 °C was observed directly via powder synchrotron XRD. As the addition of halides increases the weight of the system which is unfavourable for mobile applications, there is a strong imperative to investigate the lower chloride limits of these phases. Reducing the chloride content resulted in a mixture of rhombohedral and cubic phases except at the stoichiometry Li7(NH2)6Cl, where a single phase was observed. This new low-chloride rhombohedral phase contained little more than half of the halide present in Li4(NH2)3Cl but, importantly, maintained the enhancements seen in hydrogen desorption properties when compared to the halide-free system.
The reaction between lithium amide and magnesium chloride at 400 °C in the molar ratio 3NH2−:1Cl− resulted in the formation of a rhombohedral and/or cubic phase of stoichiometry Li3Mg0.5(NH2)3Cl, again dependent on the reaction time. Reducing the chloride content also resulted in the formation of a new low-chloride rhombohedral phase, Li6Mg0.5(NH2)6Cl. The presence of magnesium was found to reduce the desorption temperature for Li3Mg0.5(NH2)3Cl but not for Li6Mg0.5(NH2)6Cl.
The deliberate engineering at the nanoscale of compounds with much improved bulk diffusion is likely to be complementary, rather than an alternative, to current methods of kinetic enhancement, and thus affords a very high probability of producing materials whose performance is markedly superior to the current generation. These results point the way to a radical new approach for optimizing the hydrogen desorption and reabsorption kinetics that recent analysis suggests is capable of delivering a step-change in these key properties [7]. We also note that, though developed for light-weight hydrogen storage, some of these materials may have potential in a range of other energy applications such as all solid state lithium ion batteries, supercapacitors, and CO2 capture and storage membranes.
Acknowledgments
We thank the EPSRC for the studentship of Rosalind Davies via the Hydrogen, Fuel Cells and their Applications Centre for Doctoral Training, the RCUK Energy Programme (Supergen UKSHEC2) and Advantage West Midlands (Science City Hydrogen Energy Project) for funding. The Bruker D8 system used in this research was obtained, through the Science City Advanced Materials Project: Creating and Characterizing Next Generation Advanced Materials, with support from AWM and part funded by the European Regional Development Fund (ERDF). The authors would also like to acknowledge Dan Reed for his help in the preparation and running of the TPD–MS, Diamond light source for the provision of beamtime (proposal number EE3909) and Paul Adamson for help in using beamline I11.
Footnotes
- *
Invited talk at the 7th International Workshop on Advanced Materials Science and Nanotechnology IWAMSN2014, 2–6 November 2014, Ha Long, Vietnam.